Creating a pool set for matchRanges
Eric S. Davis
11/20/2025
Source:vignettes/matching_pool_set.Rmd
matching_pool_set.RmdIntroduction
matchRanges() performs subset selection on a pool of
ranges such that chosen covariates are distributionally matched to a
focal set of ranges. However, the generation of a set of pool ranges is
not a trivial task. This vignette provides some guidance for how to
generate a pool of ranges.
For this analysis, we use DNase peaks as a measure of open chromatin,
and ChIP-seq for JUN peaks as a measure of JUN binding. Suppose we are
interested in the properties of chromatin accessibility, but suspect
that JUN-binding impacts accessibility. We can use
matchRanges() to control for the DNase signal and the
length of the site so we can compare our JUN-bound sites (i.e., our
focal set) to sites where JUN is not bound (i.e., our
pool set).
Obtaining example data
First, we use AnnotationHub to access
GRanges for DNase and JUN narrowPeaks in human (hg19)
GM12878 cells:
library(AnnotationHub)
ah <- AnnotationHub()
dnase <- ah[["AH30743"]]
junPeaks <- ah[["AH22814"]]
dnase## GRanges object with 271214 ranges and 6 metadata columns:
## seqnames ranges strand | name score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] chrM 1-1096 * | Rank_1 2806
## [2] chrM 1210-4181 * | Rank_2 2806
## [3] chrM 8868-16581 * | Rank_3 2806
## [4] chrM 4661-5575 * | Rank_4 2784
## [5] chr21 9825397-9827624 * | Rank_5 2741
## ... ... ... ... . ... ...
## [271210] chr2 99101766-99101877 * | Rank_271210 20
## [271211] chr6 319032-319131 * | Rank_271211 20
## [271212] chr1 117329477-117329556 * | Rank_271212 20
## [271213] chr1 45985571-45985660 * | Rank_271213 20
## [271214] chr2 101185965-101186044 * | Rank_271214 20
## signalValue pValue qValue peak
## <numeric> <numeric> <numeric> <numeric>
## [1] 76.6491 280.660 271.178 485
## [2] 76.6491 280.660 271.178 2340
## [3] 76.6491 280.660 271.178 5029
## [4] 76.1730 278.495 271.178 288
## [5] 75.2208 274.173 268.040 814
## ... ... ... ... ...
## [271210] 2.15692 2.00390 0.46416 71
## [271211] 2.15692 2.00390 0.46416 40
## [271212] 2.19528 2.00202 0.46235 43
## [271213] 2.19528 2.00202 0.46235 42
## [271214] 2.19528 2.00202 0.46235 26
## -------
## seqinfo: 298 sequences (2 circular) from hg19 genomeSince we want to control for accessibility, we can use the
signalValue from the DNase peaks as a covariate. DNase
sites are also different lengths. If we suspect length might impact
accessibility differently at JUN-bound sites, we can include it as a
covariate as well. For visualization, let’s convert these to log-scale
using mutate() from plyranges:
Creating the focal and pool sets
Next we define our focal and pool sets. The focal set
contains the feature of interest (i.e., DNase peaks bound by JUN),
whereas the pool set lacks this feature (i.e., unbound
DNase peaks). matchRanges() is designed to handle datasets
that can be binarized into these two distinct groups. With
plyranges it is easy to filter DNase sites by overlap (or
lack of overlap) with JUN peaks:
## Define focal and pool
focal <- dnase |>
filter_by_overlaps(junPeaks)
pool <- dnase |>
filter_by_non_overlaps(junPeaks)The focal set must be smaller than the pool set for matching to work correctly. Matching is most effective when the pool set is much larger and covers all values in the focal set.
length(focal)## [1] 2490
length(pool)## [1] 268724## [1] 107.9213Before matching, the focal set shows a different distribution of length and signal than the pool set:
## Before matching, focal shows higher
## signalValue than pool
library(tidyr)
library(ggplot2)
bind_ranges(focal=focal,
pool=pool,
.id="set") |>
as.data.frame() |>
pivot_longer(cols=c("log10_length", "log10_signal"),
names_to="features",
values_to="values") |>
ggplot(aes(values, color=set)) +
facet_wrap(~features) +
stat_density(geom='line', position='identity') +
ggtitle("DNase sites") +
theme_bw() +
theme(plot.title=element_text(hjust=0.5))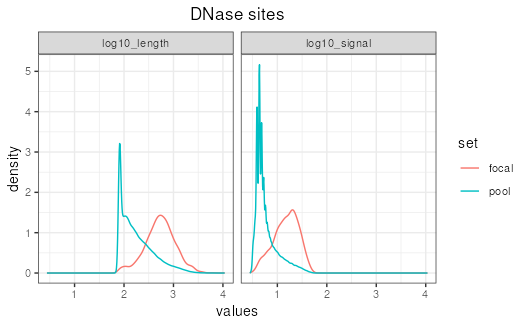
Obtaining the matched set with matchRanges()
To control for these differences, we can use
matchRanges() to select a subset of unbound DNase sites
from the pool that have the same distribution of length and signal.
library(nullranges)
set.seed(123)
mgr <- matchRanges(focal=focal,
pool=pool,
covar=~log10_length + log10_signal,
method='rejection',
replace=FALSE)
mgr## MatchedGRanges object with 2490 ranges and 8 metadata columns:
## seqnames ranges strand | name score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] chr9 140013244-140013564 * | Rank_13746 504
## [2] chr3 128839003-128841214 * | Rank_6778 736
## [3] chr7 92218841-92219942 * | Rank_8688 662
## [4] chr5 68390322-68390664 * | Rank_58053 126
## [5] chr5 43483248-43484916 * | Rank_9339 634
## ... ... ... ... . ... ...
## [2486] chr9 126889146-126889567 * | Rank_29379 272
## [2487] chr16 14723226-14724483 * | Rank_7767 695
## [2488] chr7 135627453-135627772 * | Rank_52826 142
## [2489] chr5 177643218-177643709 * | Rank_94530 73
## [2490] chr2 219433940-219435175 * | Rank_34979 223
## signalValue pValue qValue peak log10_signal log10_length
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 17.60499 50.4442 47.2874 88 1.269629 2.50786
## [2] 19.77854 73.6972 70.1244 498 1.317615 3.34498
## [3] 24.75622 66.2132 62.7729 900 1.410882 3.04258
## [4] 6.36768 12.6577 10.3340 75 0.867331 2.53656
## [5] 20.24701 63.4728 60.0874 746 1.327298 3.22272
## ... ... ... ... ... ... ...
## [2486] 11.36989 27.28862 24.58641 148 1.092366 2.62634
## [2487] 25.70838 69.58699 66.08530 924 1.426648 3.10003
## [2488] 8.09338 14.29479 11.90889 75 0.958725 2.50651
## [2489] 4.55759 7.34743 5.24424 133 0.744887 2.69285
## [2490] 9.90281 22.35572 19.75514 93 1.037538 3.09237
## -------
## seqinfo: 298 sequences (2 circular) from hg19 genomeAssessing covariate balance
Now let’s use the plotCovariate() function with
patchwork to visualize how similar our matched distribution
is to focal:
library(patchwork)
lapply(covariates(mgr),
plotCovariate,
x=mgr,
sets=c('f', 'm', 'p')) |>
Reduce('+', x=_) +
plot_layout(guides="collect") +
plot_annotation(title="DNase sites",
theme=theme(plot.title=element_text(hjust=0.40)))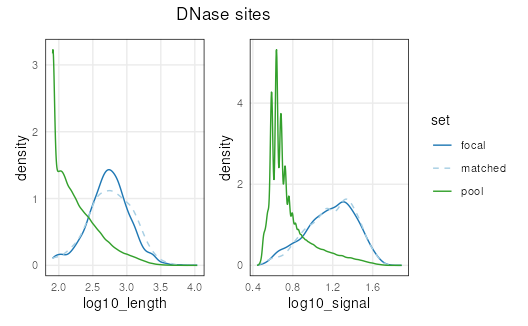
An important part of propensity-score matching, is assessing
similarity, or balance, between the focal and matched sets. One way is
to visually examine the distributions as we have done above. Another way
is to report summary statistics about the two sets. cobalt
is a package designed to specifically address covariate balance after
covariate matching. Below, we demonstrate how to use cobalt
to calculate the standardized mean differences and visualize these
statistics with a love plot. For more information about assessing
covariate balance, refer to the detailed documentation in the
cobalt vignette:
vignette("cobalt", package = "cobalt").
library(cobalt)
res <- bal.tab(f.build("set", covariates(mgr)),
data=matchedData(mgr)[!set %in% 'unmatched'],
distance="ps",
focal="focal",
which.treat="focal",
s.d.denom="all")
res## Balance by treatment pair
##
## - - - focal (0) vs. matched (1) - - -
## Balance Measures
## Type Diff.Un
## ps Distance 0.0929
## log10_length Contin. 0.1108
## log10_signal Contin. 0.0666
##
## Sample sizes
## focal matched
## All 2490 2490
##
## - - - focal (0) vs. pool (1) - - -
## Balance Measures
## Type Diff.Un
## ps Distance -1.5435
## log10_length Contin. -1.4063
## log10_signal Contin. -1.7401
##
## Sample sizes
## focal pool
## All 2490 268724
## - - - - - - - - - - - - - - - - - - - - - - - - -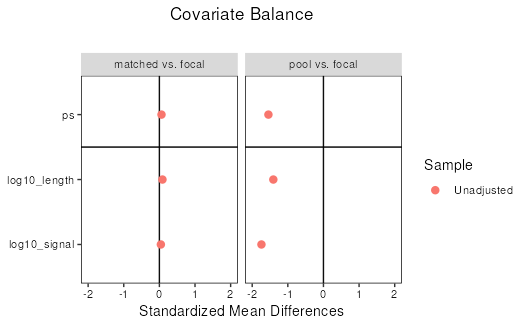
The “focal vs. matched” comparison shows much lower standardized mean differences than “focal vs. pool”, indicating that the matched set has been successfully controlled for covariates of DNAse site length and signal.